Blood Coagulation Factors and Interpretations
Blood Coagulation Factors
What sample is needed for the estimation of blood coagulation factors?
- Collect the venous blood in a blue top tube.
- It contains sodium citrate 3.8%.
- Blood to anticoagulant ratio is 9:1.
- This tube sample is used for blood clotting factor assay.
What are the precautions for blood coagulation factor assay?
- Perform the assay immediately.
- If delayed, then freeze the specimen.
- Apply the pressure bandage over the venipuncture site.
- Look for the bleeding possibility.
What are the indications for clotting factors?
- It is advised to assess the concentration of the specific coagulation factor.
How do clotting factors work for clotting?
- A cascade of coagulation factors is necessary for proper blood clotting.
- The following diagram shows the role of each coagulation factor.

Blood coagulation factors: Coagulation pathways
Blood coagulation factors:
What are the coagulation factors in the body?
- Fibrinogen (Factor 1).
- Prothrombin (Factor II).
- Thromboplastin (Factor III).
- Ionized Calcium (Factor IV).
- Proaccelerin (Factor V).
- Factor VI.
- Proconvertin (Factor VII).
- Antihemophilic factor (Factor VIII).
- Chrismats factor (Factor IX).
- Stuart factor (Factor X).
- Plasma thromboplastin antecedent (Factor XI).
- Hageman’s factor (Factor XII).
- Fibrin-stabilizing factor (Factor XIII).
Fibrinogen, Factor I:
How will you define Fibrinogen Factor 1?
- Fibrinogen is a complex protein and polypeptide, and it has an enzymatic property where it is converted into fibrin.
- Fibrinogen is a globulin protein.
- Fibrin and platelets form the blood clot to stop the bleeding.
What are the precautions for fibrinogen level estimation?
- Blood transfusion in the last month will affect the result.
- A diet containing omega-3 and omega-6 fatty acids reduces the fibrinogen level.
- Estrogen and oral contraceptives also increase the fibrinogen level.
- Anabolic agents, phenobarbitol, streptokinase, and valproic acid reduce the level of fibrinogen.
- A high level of heparin interferes with test results.
What is the role of Fibrinogen in clotting?
- Fibrinogen is necessary for the clotting mechanism.
- The liver produces fibrinogen and is also called an acute-phase protein.
- It is raised in acute inflammation and necrosis.
- When exposed to thrombin, fibrinogen splits into fibrin, forming a polymerized clot.
- This is the precursor of Fibrin.
- Fibrinogen (factor I) converts to Fibrin.
- It is part of the common pathway.
What are the causes of Fibrinogen deficiency?
- Afibrinogenemia.
- Hypofibrogenemia.
- Dysfibrogenemia.
 role in clotting")
Fibrinogen (Factor 1) role in clotting
 metabolism")
Fibrinogen (Factor 1) metabolism
What are the causes of Increased Fibrinogen levels?
- Increased level of fibrinogen is associated with:
- increased risk of coronary heart disease.
- Stroke (various cerebral accidents and diseases).
- Acute myocardial infarction.
- Peripheral arterial disease.
- Nephrotic syndrome.
- Pregnancy (eclampsia).
- Cancers.
- Multiple myeloma and Hodgkin’s lymphoma.
What are the causes of decreased Fibrinogen levels?
- A decreased level of fibrinogen is seen in:
- Liver diseases.
- Malnutrition.
- DIC (disseminated intravascular coagulopathy).
- Cancers.
- Dysfibrinogenemia.
- Primary fibrinolysis.
- Hereditary and congenital hypofibrinogenemia.
What is the normal fibrinogen level?
- Adult = 200 to 400 mg/dL (2.0 to 4.0 g/L)
- Newborn = 125 to 300 mg/dL
- Critical value = <100 mg/dL
What are the Panic values of Fibrinogen (Factor 1)?
- <50 mg/dL (<0.5 g/L) can lead to hemorrhage after traumatic surgery.
- >700 mg/dL (7.0 g/L) level is a risk for coronary artery disease and cerebrovascular disease.
Prothrombin (Factor II):
How will you define Prothrombin (Factor II)?
- Prothrombin is a glycoprotein with a molecular weight of 71,600 daltons in the blood and plasma.
- Prothrombin is a vitamin K-dependent clotting factor.
- This is produced in the liver. and needs vitamin K for its production.
What are the facts about Prothrombin (Factor II)?
- It is most abundant and has the longest half-life of the vitamin K-dependent clotting factors.
- It takes about three weeks for the body’s vitamin K stores to be exhausted.
- Prothrombin is converted to thrombin, which stimulates platelet aggregation and activates cofactors (factor X or prothrombinase), Factor C, and Factor XIII.
What is the effect of a deficiency of prothrombin (Factor II)?
- The deficiency of prothrombin will delay thrombin formation, leading to hemorrhagic symptoms.
- Hypoprothrombinemia:
- It is an autosomal recessive trait.
- This may be acquired through vitamin K deficiency or oral anticoagulant therapy, such as warfarin.
What are the signs and symptoms of Prothrombin deficiency?
- S/S depends upon the level of prothrombin.
- These patients may have H/O epistaxis, menorrhagia, post-partum hemorrhage, and hemorrhage after the surgery.
- Hemorrhages may occur after broad-spectrum antibiotic therapy.
- Prothrombin level is <2% to <50% of normal.
- It is a rare condition, and it may lead to hemorrhagic symptoms.
- PTT and PT are prolonged and have normal thrombin times.
- A definitive diagnosis depends upon the prothrombin (functional activity) assay or the prothrombin level antigenic concentration.

Role of Prothrombin in Fibrin Formation
Thromboplastin, Factor III, or Tissue factor:
How will you define Thromboplastin (Factor III)?
- This is the name given to any substance that can convert Prothrombin to Thrombin.
- Tissue thromboplastin is composed of phospholipids and lipoproteins and can be extracted from various tissues.
- The extrinsic and intrinsic pathways generate Thromboplastin.
- Thromboplastin is an enzyme that is released from damaged cells, particularly from the platelets.
- This is found in the brain, lungs, and other tissues. It is also present in the platelets.
What is the mode of action of the Thromboplastin (Factor III)?
- This is a tissue factor that will activate VII when blood is exposed to tissue fluid.
- Tissue thromboplastin forms a complex with factor VII, Ca++, and stimulates the extrinsic coagulation pathway.
- The above complex was used as a reagent for the PT test.
- It converts prothrombin to thrombin.

Thromboplastin’s role in coagulation
Ionized Calcium Factor IV:
How will you define ionized calcium?
- Ionized calcium is important for the clotting system.
- Calcium is necessary as a cofactor in several steps of the coagulation pathways.

Calcium in various forms in plasma
What are the functions of ionized calcium?
- Ionized calcium is needed in the clotting system at the following stages:
- This active form of Calcium is needed to activate thromboplastin.
- Convert Prothrombin to Thrombin.
- Activate factor XIII to XIIIa.
- Activate factor X to Xa.
- For the formation of fibrin.
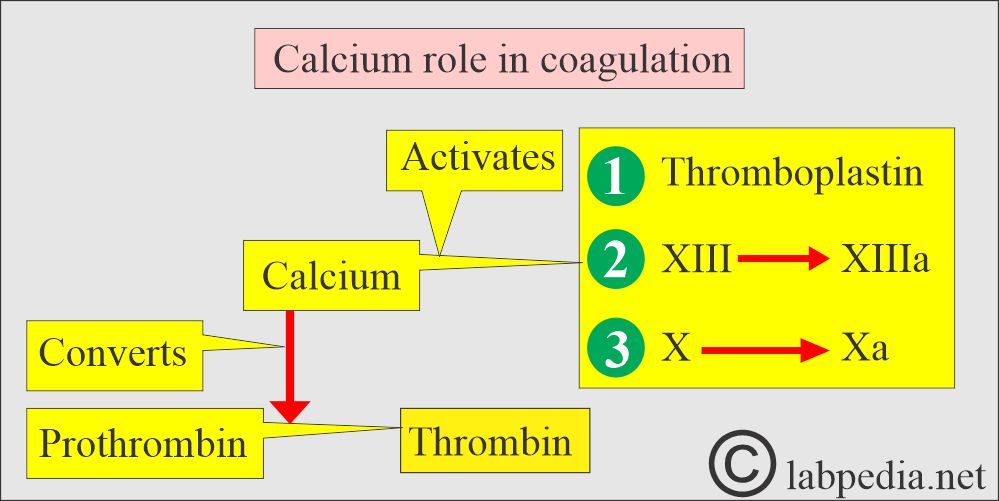
Calcium’s role in coagulation
- Calcium in the blood is approximately 50% ionized, and a very small amount is required for the clotting mechanism.
Proaccelerin, Factor V:
How will you define Proaccelerin (Factor V)?
- It is also called a labile factor.
- This is a globulin and is labile in the plasma. It is not found in the serum.
- This is synthesized in the liver; its molecular weight is 350,000 daltons, it has a short half-life, and is heat-labile.
- It is present in the α-granules of the platelets.
- This is not vitamin K-dependent.
How will you discuss the stability of factor V?
- Factor V is unstable and is reduced by 2 to 3 days when blood is refrigerated in a blood bank.
- Refrigeration preserves factor V in laboratory plasma, but even at 4°C, factor V is reduced within 24 hours.
- Freezing the specimen immediately is important to preserve factor V activity.
What are the functions of factor V?
- It is consumed during clotting and accelerates the transformation of prothrombin to thrombin.
- In the presence of factor VIII, it helps the normal coagulation process.
- This will deteriorate rapidly in oxalate plasma and slightly slower in citrated plasma.
- This is consumed in the clotting process and not found in the serum.
- This helps as a cofactor in the transfer of prothrombin to thrombin.
- Congenital deficiency of Factor V is called parahemophilia.
What are the signs and symptoms of Factor V deficiency?
- Ecchymosis.
- Epistaxis.
- Gingival bleeding.
- Gastrointestinal bleeding.
- CNS bleeding.
- Umbilical bleeding.
- Menorrhagia.
- Acquired deficiency of factor V is observed in cases of antibodies and is also associated with liver disease, carcinoma, tuberculosis, and disseminated intravascular coagulation (DIC).
What is the normal Factor V?
- Bleeding tendency is seen when this is <10%, where the normal value is 50% to 150% of normal.
- PT and APTT are prolonged in factor V deficiency.
- APTT may be normal in mild deficiency and abnormal in severe deficiency.
- Thrombin time is normal.
How will you treat Factor V deficiency?
- These patients are treated with fresh or frozen plasma.
- Cryoprecipitate does not contain an adequate amount of factor V.

Activation of coagulation factor V by Snake venom
Factor VI:
- This factor does not exist.
Proconvertin, Factor VII (Stable factor):
How will you define Proconvertin (Factor VII)?
- This factor is synthesized in the liver and depends on vitamin K for its activity.
- This is beta-globulin with a molecular weight of 50,000 daltons.
- This has a half-life of 4 to 6 hours and is produced in the liver.
- This is a vitamin K-dependent factor.
- This factor is not destroyed or consumed during the clotting process; therefore, it is found in both serum and plasma.
- This factor is activated by thromboplastin.
What are the functions of Factor VII?
- The division of the coagulation system into intrinsic and extrinsic pathways is not observed in vivo, as activated factor VIIa can activate both factors IX and X.
- Thromboplastin activates factor X.
- Its activity increases by the factor XIIa and IXa.
- Factor VII needs tissue factor, thrombin, and Ca++ to become VIIa.

Role of Factor VII
What are the causes of Factor VII deficiency?
- This may be seen in liver diseases.
- Warfarin therapy.
- Dietary vitamin K deficiency.
What are the S/S of factor VII deficiency?
- These patients develop deep muscle hematomas.
- These patients may have a joint hemorrhage.
- There may be epistaxis.
- There may be menorrhagia.
How will you diagnose factor VII deficiency?
- PT is prolonged.
- APTT is normal (factor VII is not measured in this test).
- Bleeding time is normal.
- Diagnosis of factor VII deficiency needs a one-stage factor assay.
- Patients with <1% activity may have severe bleeding manifestations (normal factor VII value is 65% to 140% of normal).
How will you treat Factor VII deficiency?
- It is treated with fresh frozen plasma.
- Vitamin K supplementation.
- Prothrombin complex concentrates.

Coagulation Factor VII activation
What are the diagnostic criteria for Factor VII deficiency?
| Tests | Normal /seconds | In the deficiency of factor VII |
|
|
|
|
|
|
|
|
|
|
|
|
Antihemophilic factor, Factor VIII:
How will you define Antihemophilic factor (Factor VIII)?
- This factor VIII was originally called antihemophilic globulin and is now known as factor VIII (anti-hemophilic factor).
- This factor is produced by the liver’s sinusoidal and endothelial cells.
- This is a glycoprotein with a molecular weight of 330,000 daltons, and it is a very important factor.
What is the site of synthesis of factor VIII?
- Its synthesis site is unclear, but the idea is that it may be the liver.
- Severe liver failure does not lead to its deficiency.
- It is considered that cells present in different organs, like fibroblasts, lymphocytes, macrophages, and vascular endothelial cells, may be a source.
What do you know about von Willibrand’s factor?
- Factor VIII is present in the plasma, is a complex with von Willebrand’s factor, and circulates in the plasma.
- von Willebrand’s factor is a glycoprotein and is synthesized by megakaryocytes and endothelial cells.
- It circulates as a complex with factor VIII, known as FVIII + vWF.
- 1% to 2% of the complex function as procoagulant (FVIII: C) can be measured by clotting assay, and the rest of the portion is FVIII:
- This factor, vWF, will mediate platelet adhesion.
How stable is Factor VIII?
- After the blood transfusion, 50% of the activity is lost in 8 to 12 hours.
- It is stable in fresh-frozen plasma.
- The lyophilization process will preserve its activity with minimal loss.
What is the importance of Factor VIII?
- A deficiency of factor VIII characterizes Hemophilia.
- This factor is consumed during clotting, so it is not found in the serum.
- FVIII: C has a half-life of 8 to 12 hours, and it acts as a cofactor.
What are the fractions of Factor VIII?
- This factor has different fractions, which are divided based on their molecular weights.
- High molecular weight type.
- A low molecular weight type.
- In hemophilia, classical factor VIII deficiency is characterized by a deficiency in the low-molecular-weight molecule, while the high-molecular-weight molecule is normal.
What are the functions of Factor VIII?
- This cofactor accelerates the conversion of factor X to Xa.
- The above cofactor converts X to Xa in the presence of factor IXa + calcium and phospholipids complex.

The role of coagulation factor VIII in the clotting
- Factor VIII is also an acute-phase protein. It will be increased in:
- Inflammation.
- Pregnancy.
- Stress.
- Infections.
Christmas factor, Factor IX:
How will you define Christmas factor (Factor IX)?
- This is a stable protein factor.
- This is a single-chain glycoprotein with a molecular weight of 60,000 daltons.
- This is synthesized in the liver and is dependent on vitamin K.
- This process is part of the intrinsic pathway, where it is activated to form XIa in the presence of Ca++.
- This is not consumed during the clotting process.
- It has a half-life of 20 hours.
- This is also not consumed by aging and is present in the serum and plasma.
- There is no significant loss of storing blood or plasma at 4 °C for 2 weeks.
What are the other names for Christmas factor (Factor IX)?
- This is also called antihemophilic factor B or Christmas factor.
What are the functions of the Chrismatis factor (Factor IX)?
- This is an essential component of the intrinsic thromboplastin generation system.
- This helps factor Va + factor VIIIa lead to their amplification.

The role of coagulation factor IX in clotting
What is the role of Factor IX in the activation of Factor X?
- It is also found that factor IXa can slowly activate factor X in the presence of phospholipids and calcium ions.
- Factor VIII alone, or along with thrombin, can not activate factor X in the absence of factor IXa.
Stuart-Prower factor, Factor X:
How will you define the Start-Prower factor (Factor X)?
- This is alpha globulin with a molecular weight of 58,800 daltons.
- It comprises light and heavy chains held together by a single disulfide bond.
- It requires Vitamin K for its synthesis in the liver and is released into plasma as a precursor to serine protease.
- Its half-life is roughly 40 hours.

Factor X structure
How is factor X activated?
- Activation of factor X to Xa involves the cleavage of a peptide bond in the heavy chain.
- This reaction in the intrinsic pathway occurs in the presence of factor VIIIa, calcium ions, and phospholipids.
- The same bond is cleaved by factor VIIa in the presence of tissue factor in the extrinsic pathway.
- This is also called thrombokinase.
- This is a relatively stable factor and is not consumed in the clotting process.
- This is found in serum and plasma.
- Factor Xa is inactivated by a serine protease inhibitor.
- This factor helps to convert prothrombin to thrombin.

Activation of coagulation factor X
How will you discuss the deficiency of factor X?
- Inherited deficiency of factor X is extremely rare. Its transmission is autosomal recessive.
- Factor X deficiency may occur at any age, but symptoms typically appear at a very young age.
What are the S/S of factor X deficiency?
- Bleeding sites vary in severity according to the deficiency.
- There may be easy bruising.
- There may be epistaxis.
- There is gastrointestinal bleeding.
- There is menorrhagia.
- In mild cases, you may see hemarthrosis.
- There may be a hemorrhage in the CNS.
- There is severe bleeding in the post-operative stage.
- Factor X deficiency has been reported in cases of amyloidosis.
How will you diagnose Factor X deficiency?
- Take family history and laboratory data.
- Workup for liver diseases and vitamin K deficiency.
| Type of the lab test | Normal value | Value in Factor X deficiency |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
How will you treat Factor X deficiency?
- It consists of fresh frozen plasma.
- Or can be treated with prothrombin-complex concentrate.
- If the deficiency is dietary, vitamin K will help to treat these cases.
Plasma thromboplastin antecedent, Factor XI:
How will you define plasma thromboplastin antecedent (Factor XI)?
- This is beta-globulin (glycoprotein). Its molecular weight is 143,000 daltons.
- It is synthesized in the liver and secreted into plasma.
- It consists of two polypeptide chains linked by single disulfide bonds.
- This is also called antihemophilic factor C (Hemophilia C).
- Deficiency of factor XI leads to hemophilia C.
- Half-life is a few hours.
What are the important features of Factor XI deficiency?
- Factor XI deficiency is an uncommon and inherited condition, characterized as an autosomal incompletely recessive trait (it affects both sexes equally).
- Deficiency varies in degree according to whether the patient is homozygous or heterozygous.
What are the signs and symptoms of Factor XI deficiency?
- The S/S are mild.
- There is mild bleeding compared to Hemophilia A and B.
- Factor XI is present in the plasma and serum and is stable in both.
- Spontaneous bleeding is rare.
- There may be bleeding after the surgery or trauma, such as dental surgery.
- How will you diagnose Factor XI deficiency?
- APTT is abnormal.
- Bleeding time and PT are normal.
What is the fate of Factor XI?
- This is partially consumed during clotting.
- This is found in the serum.
- This is needed in the intrinsic pathway to form the thromboplastin generation cascade.
- Factor XI and factor XII are “contact factors” for clotting.
- When it contacts the negatively charged surface, factor XI is activated by factor XIIa.
Hageman’s factor, Factor XII :
How will you define Hageman’s factor (Factor XII)?
- John Hageman discovered it. He found a patient with prolonged clotting time but no bleeding disorder.
- This is a single-chain β-globulin with a molecular weight of 76,000 daltons.
- It is believed to be synthesized by the liver and circulated in the blood as an inactive zymogen.
- It is a part of the intrinsic pathway.
- It is inherited as an autosomal recessive trait.
What is the mode of action of factor XII?
- This is not consumed during clotting.
- Hageman factor + glass contact converted from the inactive to the active form.
- In vitro:
- This is a surface contact factor. After contact with a negatively charged surface in vitro, such as glass, kaolin, celite, or ellagic acid, factor XII is autoactivated to XIIa. This is also converted into a serine protease.
- This process occurs within the intrinsic pathway of coagulation.
- This is activated by collagen.
- In vivo:
- Factor XII activation occurs through contact with the cell membrane and components of white blood cells (polys).
- Factor XII exposes an active site that converts prekallikrein to kallikrein and activates factor XI.
- A small amount of activated factor XIIa activates its substrates: prekallikrein, factor XI, and HMWK.
How will you discuss the deficiency of factor XII?
- It is known as the Hageman trait and is inherited in an autosomal recessive manner.
- It is not associated with clinical bleeding or hemorrhage.
- There are chances for thrombotic diseases like myocardial infarction or thromboembolism.
- These patients are asymptomatic, and there is no surgical risk for hemorrhage.
How will you diagnose Factor XII deficiency?
- The PT level is normal.
- APTT is prolonged.
- APTT is corrected by mixing with pooled normal plasma, aged serum, or adsorbed plasma.
- Factor XII assay shows decreased or absent levels, confirming the diagnosis.
What are the precautions for the collection of the blood sample for Factor XII?
- Avoid contact activation during blood collection.
- Take the blood in the plastic syringe and transfer it to siliconized anticoagulated test tubes.
- Avoid freezing or thawing.
- Perform the test in fresh plasma.
Fibrin-stabilizing factor (Fibrinase), Factor XIII:
How will you define Factor XIII?
- Factor XIII’s molecular weight is approximately 320,000 daltons, and it circulates in association with fibrinogen.
- Factor XIII has two subunits:
- The α2-chain is found in various tissues and cells, including platelets, placenta, prostate, and macrophage cells.
- The β2-subunit is synthesized in the liver and circulates in the blood as a free dimer.
- It is postulated that the β2-subunit helps in the stabilization of the α2-subunit.
- The function of the β2-subunit is still not clear.
- This Factor XIII acts in the presence of Calcium.
What are the important facts about Factor XIII?
- This is a serum factor.
- Adding Ca++ and serum factor (Factor XIII) gives rise to coarse fibrin clot formation.
- This stabilizes polymerized fibrin monomer in the initial stage of clot formation.

Factor XIII’s role in the stabilization of fibrin for a stable clot
What is the mode of action of factor XIII?
- In the final stage of the coagulation process, there are:
- Generation of thrombin.
- Polymerization of the fibrin.
- Activation of factor XIII, which is responsible for a stable fibrin clot.
- Factor XIII is a proenzyme for plasma transglutaminase.
- In the presence of fibrin, thrombin converts Factor XIII to an enzyme called Factor XIIIa.
What are the functions of Factor XIII?
- It stabilizes the fibrin clot.
- It acts as a catalyst, forming a bond between various proteins, like:
- Fibrin monomer.
- Fibronectin.
- Collagenase.
- α2-plasmin inhibitor.

Factor XIII deficiency
- It leads to the Cross-linking of various proteins, which leads to:
- Hemostasis.
- Maintenance of pregnancy.
- Wound healing.
How will you describe the deficiency of factor XIII?
- It may be a congenital deficiency as an autosomal recessive trait.
- It may also be a homozygous deficiency.
What are the S/S of factor XIII deficiency?
- In homozygous cases, there are moderate to severe attacks of hemorrhage.
- In typical cases, there is an initial stoppage of the bleeding followed by a recurrence of the bleeding 36 hours or more after the initial trauma.
- This repeated episode of bleeding is due to the dissolution of the fibrin clot and is not stabilized by factor XIII deficiency.
- At birth, there is bleeding from the umbilicus.
- Acquired partial deficiency of factor XIII is seen in leukemias, severe liver disease, and DIC cases.

Factor XIII deficiency
How will you diagnose Factor XIII deficiency?
- PT, APTT, fibrinogen level, bleeding time (BT), and platelet counts are normal.
- Screening test for factor XIII deficiency:
- Solubility of a recalcified plasma clot in a 5-molar urea solution. The clot will dissolve within 24 hours.
What are the normal values of clotting factors?
| Factors | Normal value Source 1 | Normal value Source 2 | Normal value Source 3 |
|
|
|
|
| Quantitation of the minimum hemostatic level mg/dL | Plasma concentration mg/dL | ||
|
|
|
|
|
|||
|
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|